กลุ่มอาการเทรชเชอร์ คอลลินส์ (Treacher Collins syndrome)
นพ.นนท์ โรจน์วชิรนนท์
9 ก.ค. 2560
โรคนี้พบได้ไม่บ่อยเลย เพียง 1 ต่อ 50,000 คนของเด็กเกิดมีชีพ จัดอยู่ในกลุ่มโรคที่มีการเจริญเติบโตผิดปกติของใบหน้าและขากรรไกรล่าง (mandibulofacial dysostosis หรือ MFD) โดยส่วนใหญ่พบมีการกลายพันธุ์ของยีนต้นเหตุของโรคโดยไม่พบมีใครในครอบครัวเป็นโรคเดียวกัน แสดงว่าเป็นจากปัจจัยภายนอกหรือสิ่งแวดล้อม ไม่ได้รับการถ่ายทอดมาจากพ่อแม่เสมอไป แต่ตัวผู้ป่วยเองจะถ่ายทอดโรคต่อไปยังลูกหลานได้
การกลายพันธุ์ของยีนที่ว่าเกิดขึ้นในช่วงที่ตัวอ่อนในครรภ์มีอายุ 5-6 สัปดาห์ ทำให้ส่วนโค้งแบรงเคียล (branchial arch) ที่ 1 และ 2 ทั้งสองข้างไม่สามารถเจริญเติบโตไปเป็นส่วนต่างๆ บนใบหน้าตามปกติ โดยเฉพาะอย่างยิ่งกระดูกโหนกแก้ม ขากรรไกรล่าง ใบหูและหูชั้นกลาง
การวินิจฉัย
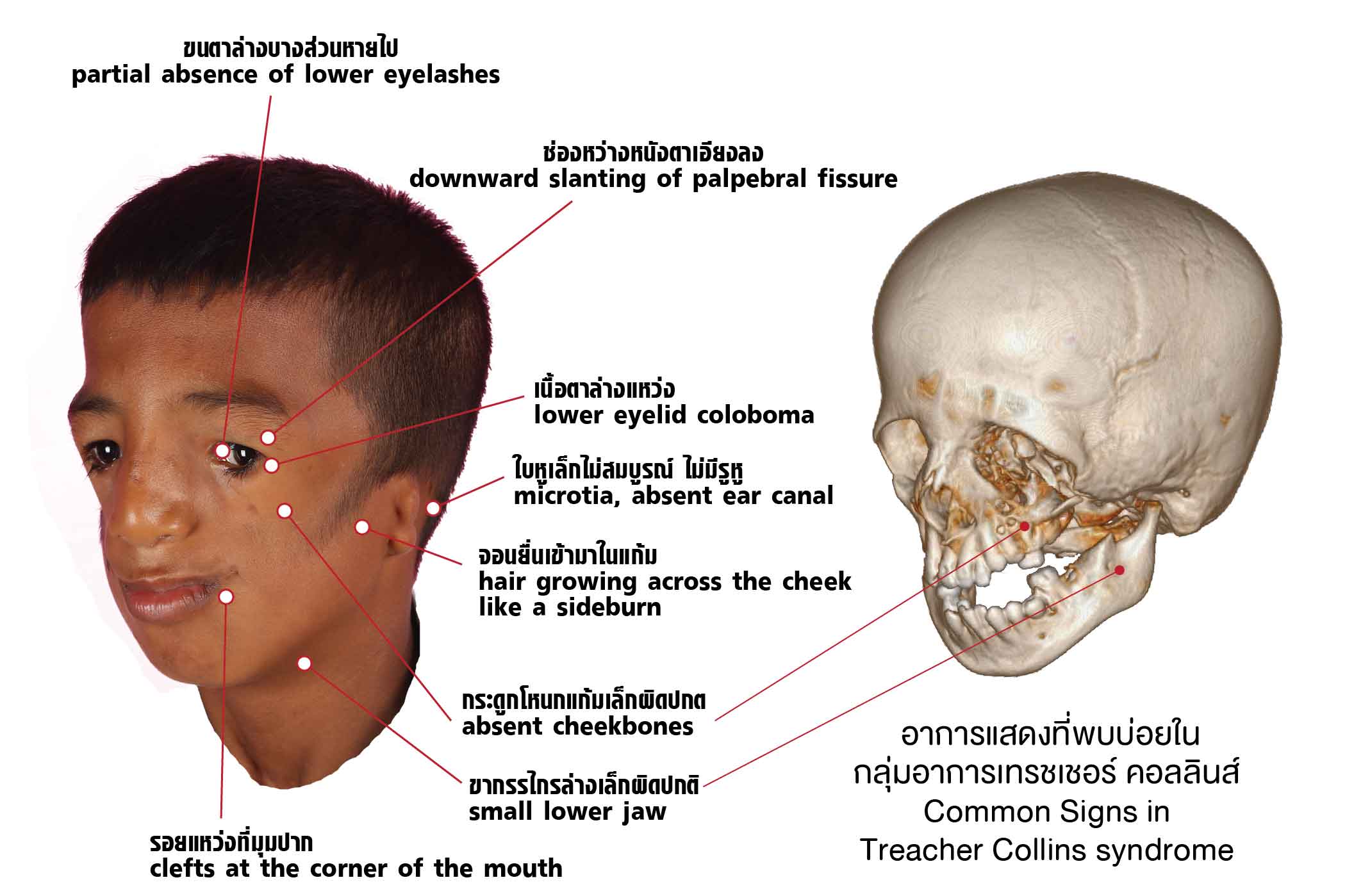
ลักษณะเด่นของโรคนี้ คือ ใบหน้าส่วนที่พัฒนามาจากส่วนโค้งแบรงเคียล (branchial arch) ที่ 1 และ 2 ตอนยังเป็นลูกอ่อนครรภ์มีความพิการที่พิเศษเฉพาะเหมือนๆ กันทั้งสองข้าง เรียกว่า เห็นปุ๊บวินิจฉัยได้ปั๊บ
- โหนกแก้มเล็กผิดปกติ
- แนวผมบริเวณขมับต่ำและจอนยื่นเข้ามาในแก้ม
- ขากรรไกรล่างเล็กผิดปกติ
- รอยแหว่งที่มุมปาก ทำให้ปากดูกว้างกว่าปกติ
- ความผิดปกติของหู
- ใบหูผิดรูป
- การนำเสียงบกพร่อง จากการตีบตันช่องหูชั้นนอกและหูชั้นกลางไม่สมบูรณ์
- มีติ่งเนื้อบริเวณหู
- ความผิดปกติของตา
- ช่องหว่างหนังตาเอียงลง
- เนื้อตาล่างแหว่ง
- ขนตาล่างบางส่วนหายไป
ยังมีโชคดีประการหนึ่ง คือ โรคนี้ตั้งแต่แรกเกิดจากโตเป็นผู้ใหญ่มีความผิดปกติของใบหน้าไม่เปลี่ยนแปลง ไม่รุนแรงมากขึ้นแต่ก็ไม่ลดน้อยลง
การรักษา
การรักษาหลักสำหรับโรคนี้คือการผ่าตัดให้รูปหน้าใกล้เคียงปกติที่สุด แต่ในปัจจุบันความก้าวหน้าทางวิทยาการทำให้สามารถวินิจฉัยโรคนี้ได้ตั้งแต่ตั้งครรภ์ ประกอบมีความรู้มากขึ้นเกี่ยวกับกลไกการเกิดโรค กำลังมีการศึกษาทดลองในสัตว์ พยายามป้องกันการเกิดความพิการตั้งแต่ในท้อง ในอนาคตเราอาจสามารถใช้ยาหรือปรับเปลี่ยนยีนเพื่อป้องกันการเกิดความผิดปกติได้ตั้งแต่อยู่ในครรภ์
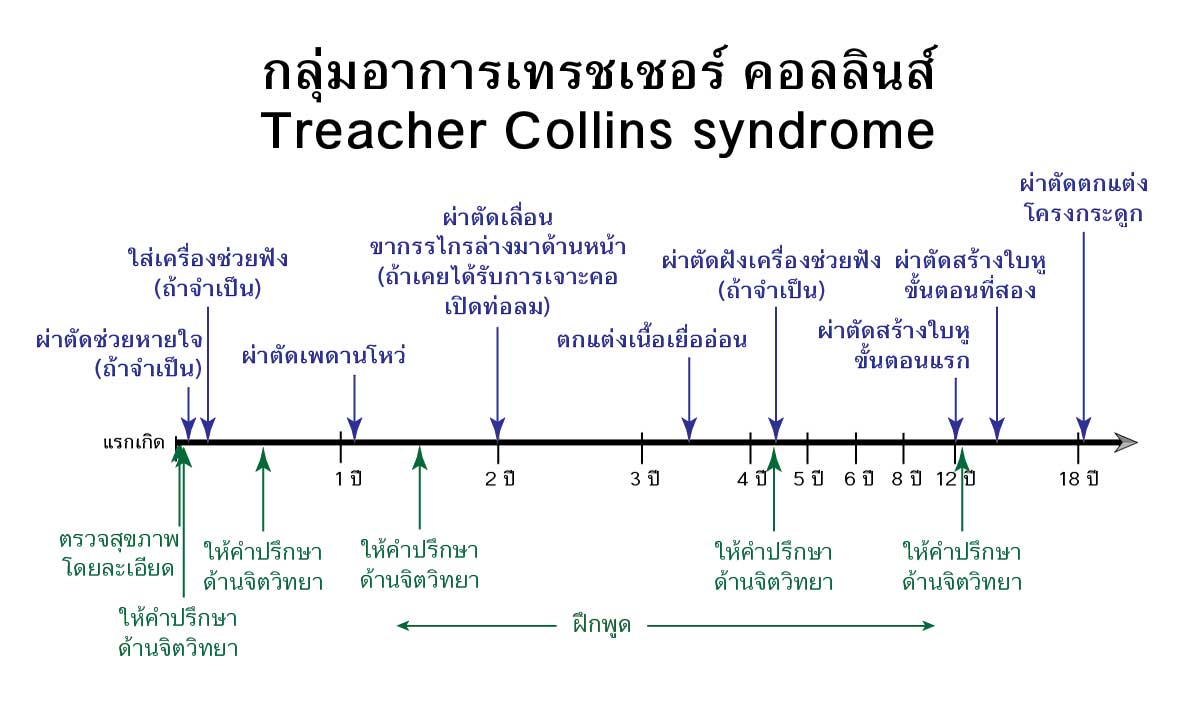
การรักษาปัญหาซ่อนเร้นและภาวะฉุกเฉินหรือเร่งด่วน
โรคนี้ไม่ได้มีแค่หน้าตาที่แปลกประหลาด แต่ยังมีปัญหาสำคัญที่เป็นอันตรายหรือวินิจฉัยไม่ได้อยู่หลายประการ ตั้งแต่แรกเกิดจนตลอดช่วงวัยทารก จึงจำเป็นต้องมีการประเมินเบื้องต้นในประเด็นต่างๆ ดังต่อไปนี้
ภาวะสูญเสียการได้ยิน
ส่วนใหญ่ของคนไข้มีปัญหาด้านการได้ยิน โดยเฉพาะอย่างยิ่งการนำเสียงบกพร่องเนื่องจากความผิดปกติของหูชั้นนอกและหูชั้นกลาง ซึ่งพัฒนามาจากส่วนโค้งแบรงเคียลที่ 1 และ 2 ที่มีปัญหา จำเป็นที่จะต้องประเมินการได้ยินและจัดหาเครื่องช่วยฟังตั้งแต่แรกเกิด เพื่อให้ผู้ป่วยได้มีพัฒนาการด้านภาษาเป็นปกติ ซึ่งยิ่งเริ่มได้เร็ว เด็กก็จะยอมรับยอมใช้เครื่องช่วยฟังได้ง่ายกว่า โดยที่พ่อแม่จะต้องคอยดูแลเครื่องช่วยฟังให้อยู่ในสภาพที่ดี
ภาวะทางเดินหายใจอุดตัน
พบได้เกือบหนึ่งในสี่ของผู้ป่วย สาเหตุส่วนใหญ่เนื่องจากกรามล่างที่เล็กมากๆ ในขณะที่ลิ้นมีการเจริญเติบโตตามปกติ ทำให้ขนาดลิ้นใหญ่เมื่อเทียบกับขนาดช่องปากและอุดตันช่องทางการหายใจ การตีบแคบของทางเดินหายใจอาจรุนแรงมากจนผู้ป่วยมีอาการหายใจลำบากตั้งแต่วัยทารก ขาดออกซิเจนต้องได้รับการรักษาอย่างเร่งด่วน
การรักษาที่ทำได้ เช่น จัดท่านอนตะแคงหรือกึ่งคว่ำ การใช้เครื่องอัดอากาศขณะหายใจเข้า การให้อากาศและหรือออกซิเจนในรูปแบบต่างๆ การใส่ท่อหายใจทางจมูก การใส่ท่อทางเดินหายใจ การผ่าตัดศัลยกรรมทำรูเปิดท่อลมหรือที่นิยมเรียกว่า การเจาะคอ การผ่าตัดยืดขยายกระดูกกรามล่าง (distraction osteogenesis) ออกมาทางด้านหน้า
เพดานโหว่
เพดานโหว่พบได้เกือบหนึ่งในสี่ของผู้ป่วย และมักจะมีปัญหาการระบายของหูชั้นกลางด้วย เกิดน้ำคั่งในหูชั้นกลางและการติดเชื้อ ซึ่งถ้าไม่ได้รับการรักษาจะมีปัญหาพูดไม่ชัดจากเพดานโหว่และหูตึงจากปัญหาหูชั้นกลาง แต่เวลาที่เหมาะสมสำหรับการผ่าตัดปิดเพดานโหว่จะล่าช้ากว่าผู้ที่มีเพดานโหว่โดยมีกรามล่างปกติ การผ่าตัดปิดเพดานโหว่จะทำเมื่อแน่ใจแล้วว่า จะมีช่องทางหายใจเพียงพอถ้าเพดานที่โหว่ถูกเย็บปิดแล้ว
โรคหัวใจแต่กำเนิด
พบได้ราวร้อยละ 10 ของผู้ป่วย จำเป็นต้องตรวจรักษาให้ดีก่อนจะมีการผ่าตัดอื่นๆ
การรักษาเฉพาะ
ช่วงวัยอนุบาลหรือวัยก่อนเรียน จนถึงช่วงวัยประถมหรือวัยเรียน
นอกจากการประเมินและแก้ไขปัญหาที่รุนแรงฉุกเฉินหรือซ่อนเร้นอยู่อย่างเหมาะสมแล้ว ผู้ป่วยแข็งแรงดีแล้ว จึงจะพิจารณารักษาผ่าตัดความพิการผิดรูปของเนื้อเยื่ออ่อนบนใบหน้า เช่น รอยแหว่งที่มุมปาก ใบหูผิดรูป (แบบที่ไม่ใช่ใบหูเล็กไม่สมบูรณ์) ติ่งเนื้อบริเวณหู เนื้อตาล่างแหว่ง ต่อไป
ควรมีการประเมินดูแลรักษาอวัยวะต่างๆ บนใบหน้าให้มีการทำงานเป็นปกติมากที่สุด โดยทำอย่างต่อเนื่อง ทั้งการได้ยิน การฝึกพูดถ้ามีเพดานโหว่ การหายใจ และการดูแลสุขภาพฟันและเหงือก ที่สำคัญต้องไม่ลืมที่จะดูแลด้านจิตใจให้ดีอัน เนื่องจากใบหน้ามักมีความแปลกแตกต่างจากเด็กปกติ พ่อแม่ต้องมีความรู้ในเรื่องของการเลี้ยงดูลูกอย่างถูกต้องตั้งแต่วัยก่อนเรียน ให้ความสำคัญกับคุณลักษณะภายในมากกว่ารูปลักษณ์ภายนอก พบปะปรึกษานักจิตวิทยาอย่างสม่ำเสมอ เพื่อป้องกันปัญหาจากการถูกล้อเลียนและสูญเสียความนับถือตัวเองเมื่อโตขึ้น ดีกว่าการตามแก้ปัญหาเมื่อเกิดขึ้นแล้วในภายหลัง
ช่วงวัยรุ่น
ช่วงนี้จะมีการผ่าตัดใหญ่เพื่อปรับเปลี่ยนโครงกระดูกใบหน้า
- ใบหูเล็กไม่สมบูรณ์ (microtia) ช่วงนี้เหมาะกับการสร้างใบหูซึ่งต้องทำอย่างน้อยสองครั้งในช่วงเวลาหลายปี
- ต้องรักษาสุขภาพฟันและเหงือกให้ดีให้พร้อมสำหรับการจัดฟันและผ่าตัดขากรรไกรบน-ล่างที่จะเกิดขึ้นหลังอายุ 16-18 ปี
- การผ่าตัดเสริมโหนกแก้มให้มีขนาดรูปร่างกลับมาเป็นปกติ ซึ่งมีหลายวิธี เช่น ตัดเลื่อนกระดูก ฉีดไขมัน ปลูกกระตูกตนเอง ใช้วัสดุสังเคราะห์เสริม แก้ไขช่องหว่างหนังตาเอียงลง
บทสรุป
โรคนี้วินิจฉัยได้ง่ายเนื่องจากลักษณะใบหน้าที่สังเกตจดจำได้ง่าย ปัจจุบันเรารู้จักโรคนี้ดีขึ้นมาก รู้จักยีนที่มีการกลายพันธุ์จนทำให้เกิดโรค การผ่าตัดแก้ไขความพิการบนใบหน้าก็ได้ผลดีขึ้นเรื่อยๆ โดยสิ่งที่นักวิจัยสนใจตอนนี้คือการศึกษายีนในระดับโมเลกุลและการป้องกันการเกิดความพิการตั้งแต่ในครรภ์